Chemical Synthesis

Over twenty-one million chemical compounds were known as of 2003. Most have been synthesized by chemists; only a small fraction of these are compounds isolated from natural sources. The final proof of a naturally occurring compound's structure is established by synthesizing the compound from simpler molecules by means of identifiable and reproducible reactions.
Protecting Groups
When synthesizing a molecule with more than one functional group, it may be difficult to carry out a reaction with one group without unintentionally
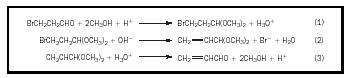
interfering or reacting with another group. Use of a protecting group helps to prevent this. The protecting group must be removed after the desired reaction is completed. In the simple example shown in Figure 1, HBr could not be eliminated from the bromopropionaldehyde because the aldehyde group would react with the base. Protecting the aldehyde group by converting it to an acetal allows the HBr elimination to take place. Other examples can be noted: alcohols or phenols can be converted to esters or ethers, aldehydes or ketones to acetals, carboxylic acids to esters, and amino groups to amides.
Retrosynthetic Analysis
For synthesis of fairly complicated molecules, the concept of retrosynthetic analysis (also called the disconnection approach), stated formally as a principle by American chemist E. J. Corey, is generally employed. In this approach, the molecule is broken up into two or more parts called synthons. A symbol used to indicate a retrosynthetic step is an open arrow written from product to suitable precursors or fragments of those precursors (Figure 2). Each synthon is similarly broken up and the process repeated until the fragments are available starting molecules. The synthesis is essentially worked backwards to the actual process followed in the laboratory. Beginning in the 1960s the strategy of organic synthesis became sufficiently systematic that computers could be used for syntheses planning.
Some of the better-known compounds synthesized by retrosynthetic analysis are strychnine, penicillin, prostaglandins, progesterone, vitamin B12, biotin, L-hexoses, menthol, and taxol.
Coordination Compounds
A coordination compound or complex refers to the grouping that is formed when a metal ion or atom accepts a pair of electrons from a molecule or ion. Metal ions—even in very low concentrations—function as powerful catalysts in many important industrial organic processes, as well as with enzymes (catalysts in living tissues). The total number of electron-donor atoms or donor pairs bonded to a given metal atom or metal cation is referred to as the coordination number. The coordination number of a compound can range from two to twelve and determines geometrical shape and physical properties.
Coordination number zero corresponds to an isolated atom; coordination number one occurs for very simple molecule combinations such as Ni-NN that are stable only in very cold matrices such as argon. Coordination
number three is fairly rare since the metal can still serve as acceptor to more Lewis bases. Coordination number four refers to the smallest number of ligands commonly found in transition metal complexes. Coordination number five was thought to be nonexistent or rare at one time; more recently, studies have revealed stable five-coordinate complexes. Most transition metal complexes have a coordination number of six and show octahedral geometry, although other geometry is possible. Coordination numbers greater than six are rare because of high ligand-ligand steric repulsion. Only small ligand atoms such as fluorine (F) and oxygen (O) have low enough repulsion to form stable seven-coordinate complexes. For coordination number eight, the number of examples is limited because of high ligand-ligand repulsion; most examples involve small ligand atoms. However, eight is a relatively favorable number for complexes of f-block elements (the lanthanides and actinides), since these are large in size and also have a larger number of valence orbitals. Higher coordination numbers of nine and ten are known that also involve f-block elements.
In 1937 English chemist Nevil V. Sidgwick suggested a rule (the octet rule for first-row p-block elements) for complex formation under which a metal can acquire ligands until the total number of electrons around it is equal to the number surrounding the next noble gas. This rule was later expanded as the eighteen-electron rule under which a d-block transition metal atom has eighteen electrons in its nine valence orbitals [five n d; one (n + 1) s, and three (n + 1) p] and will form the stablest compounds when engaged in nine bonding molecular orbitals containing eighteen electrons.
Catalysts
Catalytic processes abound in nature. From enzymes to mineral surfaces, catalysts increase the rate of a given reaction, often by reducing the activation energy that the reactants must overcome before they go on to form products. Catalysts have been developed for a wide spectrum of reactions; a common example is the catalytic converter, used in cars to reduce toxic emissions. Inexpensive transportation fuels, high-temperature lubricants, chlorine-free refrigerants, high-strength polymers, stain-resistant fibers, cancer treatment drugs, and many thousands of other products would not be possible without the existence of catalysts. Catalysts are also essential for the reduction of air and water pollution, contributing to the reduction of product emissions that are harmful to human health and the environment.
Most catalysts can be described as either homogeneous or heterogeneous . Homogeneous catalysts are molecularly dispersed with the reactants in the same phase, which provides easy access to the catalytic site but can make the separation of catalyst and products difficult. Heterogeneous catalysts—usually solids—are in a different phase from the reactants, reducing separation problems but providing more limited access to the catalytic site. Approaches to dealing with these disparate properties include anchoring the catalyst to a soluble or insoluble support, effectively "heterogenizing" the catalyst, or designing the catalyst so that it is soluble in a solvent that, under some conditions, does not mix with the reaction product. Some reactions will not take place (or will take place at a slower rate) without a catalyst being present. Actually an intermediate reaction of one of the reagents with

the catalyst (or catalyst surface) takes place at a faster rate than without it being present.
Enantiomers. Different enantiomers (mirror image forms) of a given biomolecule can exhibit dramatically different biological activities. Enzymes have evolved to catalyze reactions with selectivity for the formation of one enantiomeric form over the other. Chemists have developed various synthetic small-molecule catalysts that can achieve levels of selectivity approaching (and in some cases matching) those observed in enzymatic reactions. American chemist William S. Knowles pointed out in his 2001 Nobel Prize address that the best synthetic catalysts demonstrate useful levels of enantioselectivity for a wide range of substrates. Such catalysts have been called "privileged chiral catalysts." Such generality of scope is not observed in enzymatic catalysis.
SEE ALSO Acid-Base Chemistry ; Catalysis and Catalysts ; Coordination Compounds ; Inorganic Chemistry ; Organic Chemistry .
A. G. Pinkus
Bibliography
Basolo, Fred, and Pearson, Ralph G. (1967). "The Theory of the Coordinate Bond" (Chapter 2) and "Metal Ion Catalysis of Organic Reactions" (Chapter 8). In Mechanisms of Inorganic Reactions: A Study of Chemical Complexes in Solution, 2nd edition. New York: Wiley.
"Catalysis" (2003). Science 299:1,683–1,706.
Corey, E. J., and Chen, Xue-Min (1989). The Logic of Chemical Synthesis. New York: Wiley.
Cotton, F. Albert, and Wilkinson, Geoffrey (1980). Advanced Inorganic Chemistry: A Comprehensive Text. New York: Wiley.
Greene, T. W., and Wuts, P. G. M. (1991). Protective Groups in Organic Synthesis, 2nd edition. New York: Wiley-Interscience.
Hanson, James R. (1999). Protecting Groups in Organic Chemistry. Sheffield, England: Blackwell.
House, James E., and House, Kathleen A. (2001). "Structure and Bonding in Coordination Compounds" (Chapter 19) and "Synthesis and Reactions of Coordination Compounds" (Chapter 20). In Descriptive Inorganic Chemistry. San Diego: Harcourt/Academic Press.
Knowles, W. S. (2002). "Asymmetric Hydrogenations." Angewandte Chemie International Edition 41(12): 1,998–2,007.
Nicolaou, K. C., and Sorensen, E. J. (1996). Classics in Total Synthesis: Targets, Strategies, Methods. Weinheim, NY: VHF.
Porterfield, William W. (1993). Inorganic Chemistry A Unified Approach. San Diego: Academic Press.
Solomons, T. W. Graham (1992). Organic Chemistry, 5th edition. New York: Wiley.
Warren, Stuart (1982). Organic Synthesis: The Disconnection Approach. New York: Wiley.
Comment about this article, ask questions, or add new information about this topic: